accuracy-benchmarking
accuracy-benchmarking.RmdThis vignette is designed to validate the accuracy of the key functions from the SNPfiltR package against the established VCFtools program. For each function, I filtered 3 vcf files according to the same parameter thresholds using SNPfiltR and VCFtools, and then tested whether each genotype in the output vcf files matched. All functions matched exactly, except for the distance_thin() function. It seems that the SNPfiltR approach accurately thinned the dataset, while the ‘–thin 100’ flag from VCFtools allowed SNPs to remain in the output dataset despite being within 100 base pairs of nearby SNPs. I am not sure why VCFtools is less accurate on these vcf files I used for testing, and encourage more testing from other users in case this is an idiosyncratic issue with the particular vcf files I used. Meanwhile, I suggest using the SNPfiltR function distance_thin() which is validated clearly here, over the VCFtools approach.
All of the input vcf files necessary to fully reproduce this accuracy comparison can be found here
library(SNPfiltR)## This is SNPfiltR v.1.0.1
##
## Detailed usage information is available at: devonderaad.github.io/SNPfiltR/
##
## If you use SNPfiltR in your published work, please cite the following papers:
##
## DeRaad, D.A. (2022), SNPfiltR: an R package for interactive and reproducible SNP filtering. Molecular Ecology Resources, 00, 1-15. http://doi.org/10.1111/1755-0998.13618
##
## Knaus, Brian J., and Niklaus J. Grunwald. 2017. VCFR: a package to manipulate and visualize variant call format data in R. Molecular Ecology Resources, 17.1:44-53. http://doi.org/10.1111/1755-0998.12549##
## ***** *** vcfR *** *****
## This is vcfR 1.14.0
## browseVignettes('vcfR') # Documentation
## citation('vcfR') # Citation
## ***** ***** ***** *****Validate the hard_filter() function
Filter each vcf with SNPfiltR
#perform filtering on the 10K SNP vcf file and save the result
r.10k<-hard_filter(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.10K.vcf.gz"),depth = 5,gq = 30)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 10000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 10000
## Character matrix gt cols: 109
## skip: 0
## nrows: 10000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant: 10000
## All variants processed## 32.58% of genotypes fall below a read depth of 5 and were converted to NA## 1.36% of genotypes fall below a genotype quality of 30 and were converted to NA
r.20k<-hard_filter(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.20K.vcf.gz"),depth = 5,gq = 30)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 20000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 20000
## Character matrix gt cols: 109
## skip: 0
## nrows: 20000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant: 20000
## All variants processed## 30.09% of genotypes fall below a read depth of 5 and were converted to NA## 1.43% of genotypes fall below a genotype quality of 30 and were converted to NA
r.50k<-hard_filter(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.50K.vcf.gz"),depth = 5,gq = 30)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 50000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 50000
## Character matrix gt cols: 109
## skip: 0
## nrows: 50000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant 21000
Processed variant 22000
Processed variant 23000
Processed variant 24000
Processed variant 25000
Processed variant 26000
Processed variant 27000
Processed variant 28000
Processed variant 29000
Processed variant 30000
Processed variant 31000
Processed variant 32000
Processed variant 33000
Processed variant 34000
Processed variant 35000
Processed variant 36000
Processed variant 37000
Processed variant 38000
Processed variant 39000
Processed variant 40000
Processed variant 41000
Processed variant 42000
Processed variant 43000
Processed variant 44000
Processed variant 45000
Processed variant 46000
Processed variant 47000
Processed variant 48000
Processed variant 49000
Processed variant 50000
Processed variant: 50000
## All variants processed## 31.18% of genotypes fall below a read depth of 5 and were converted to NA## 1.37% of genotypes fall below a genotype quality of 30 and were converted to NAFilter each with VCFtools
cd /Users/devder/Desktop/benchmarking.vcfs
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.10K.vcf.gz --minGQ 30 --minDP 5 --recode --recode-INFO-all --out 10K.filtered
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.20K.vcf.gz --minGQ 30 --minDP 5 --recode --recode-INFO-all --out 20K.filtered
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.50K.vcf.gz --minGQ 30 --minDP 5 --recode --recode-INFO-all --out 50K.filtered##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.10K.vcf.gz
## --recode-INFO-all
## --minDP 5
## --minGQ 30
## --out 10K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 10000 out of a possible 10000 Sites
## Run Time = 1.00 seconds
##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.20K.vcf.gz
## --recode-INFO-all
## --minDP 5
## --minGQ 30
## --out 20K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 20000 out of a possible 20000 Sites
## Run Time = 2.00 seconds
##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.50K.vcf.gz
## --recode-INFO-all
## --minDP 5
## --minGQ 30
## --out 50K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 50000 out of a possible 50000 Sites
## Run Time = 5.00 secondsRead in VCFtools files
#read in all the vcftools files
bash.10k<-read.vcfR("~/Desktop/benchmarking.vcfs/10k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 10000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 10000
## Character matrix gt cols: 109
## skip: 0
## nrows: 10000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant: 10000
## All variants processed
bash.20k<-read.vcfR("~/Desktop/benchmarking.vcfs/20k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 20000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 20000
## Character matrix gt cols: 109
## skip: 0
## nrows: 20000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant: 20000
## All variants processed
bash.50k<-read.vcfR("~/Desktop/benchmarking.vcfs/50k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 50000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 50000
## Character matrix gt cols: 109
## skip: 0
## nrows: 50000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant 21000
Processed variant 22000
Processed variant 23000
Processed variant 24000
Processed variant 25000
Processed variant 26000
Processed variant 27000
Processed variant 28000
Processed variant 29000
Processed variant 30000
Processed variant 31000
Processed variant 32000
Processed variant 33000
Processed variant 34000
Processed variant 35000
Processed variant 36000
Processed variant 37000
Processed variant 38000
Processed variant 39000
Processed variant 40000
Processed variant 41000
Processed variant 42000
Processed variant 43000
Processed variant 44000
Processed variant 45000
Processed variant 46000
Processed variant 47000
Processed variant 48000
Processed variant 49000
Processed variant 50000
Processed variant: 50000
## All variants processedValidate the missing_by_snp() function
Filter each vcf with SNPfiltR
#perform filtering on each vcf file and save the result
r.10k<-missing_by_snp(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.10K.vcf.gz"),cutoff=.5)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 10000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 10000
## Character matrix gt cols: 109
## skip: 0
## nrows: 10000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant: 10000
## All variants processed## cutoff is specified, filtered vcfR object will be returned## 85.65% of SNPs fell below a completeness cutoff of 0.5 and were removed from the VCF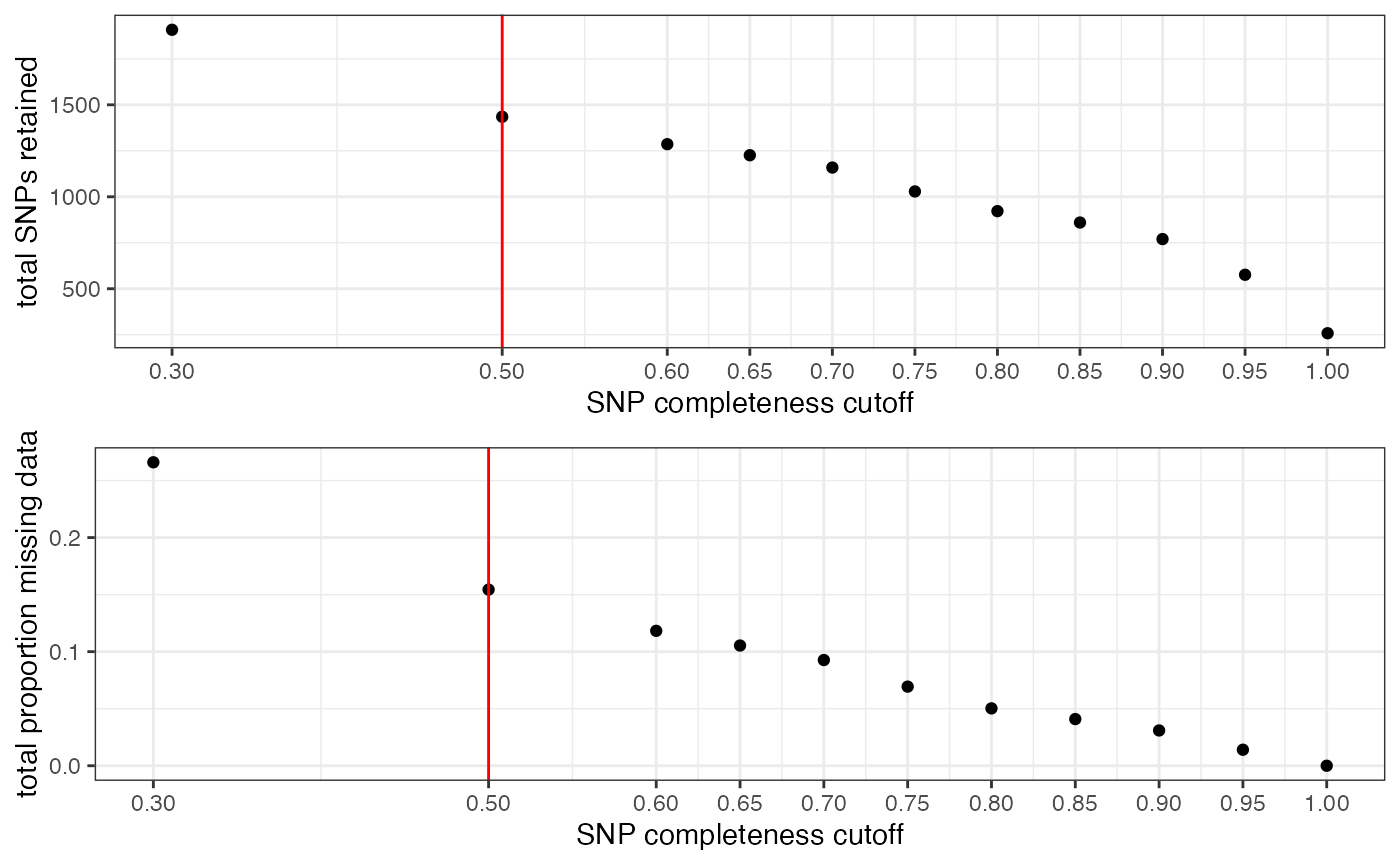
r.20k<-missing_by_snp(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.20K.vcf.gz"),cutoff=.5)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 20000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 20000
## Character matrix gt cols: 109
## skip: 0
## nrows: 20000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant: 20000
## All variants processed## cutoff is specified, filtered vcfR object will be returned## 83.8% of SNPs fell below a completeness cutoff of 0.5 and were removed from the VCF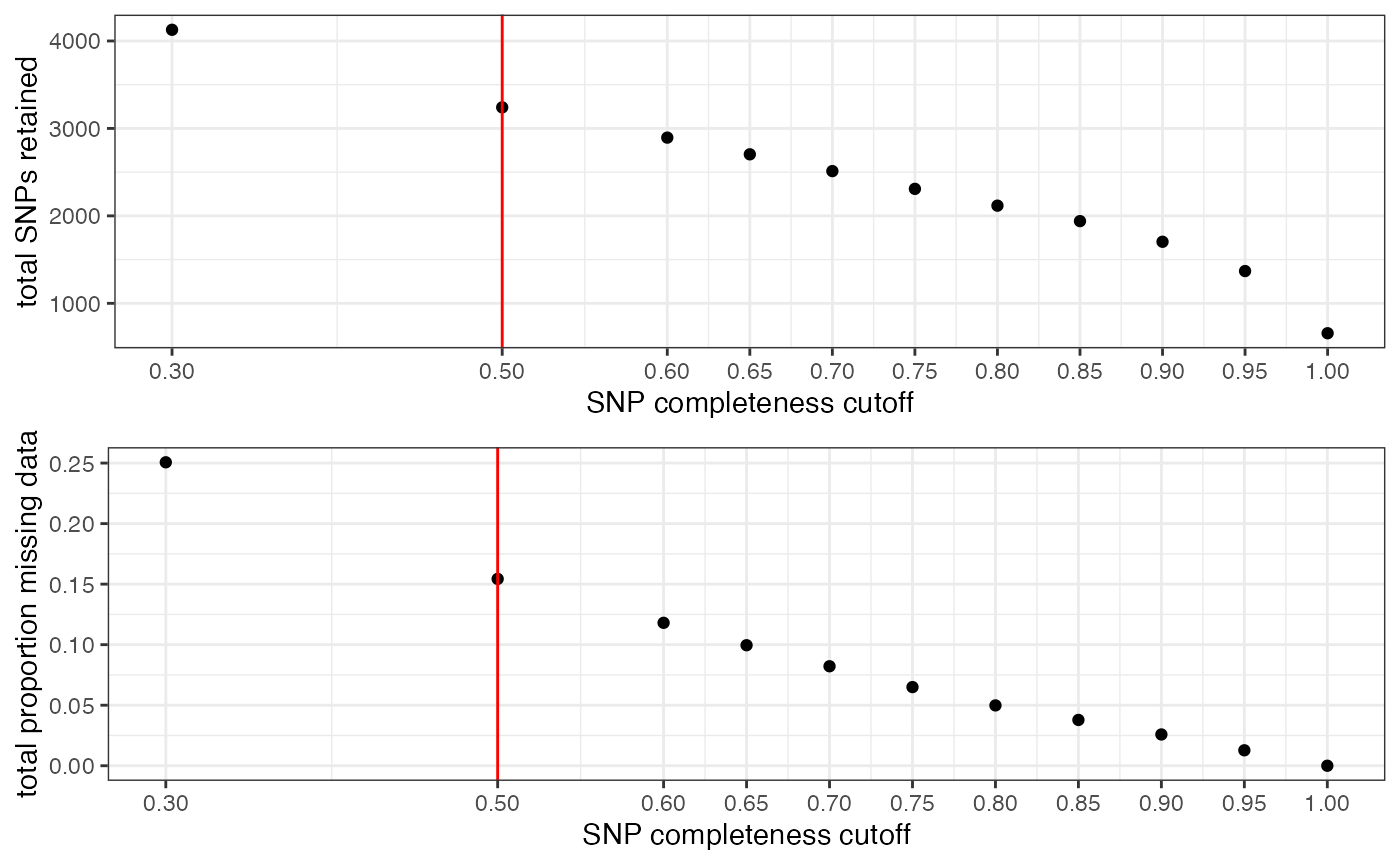
r.50k<-missing_by_snp(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.50K.vcf.gz"),cutoff=.5)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 50000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 50000
## Character matrix gt cols: 109
## skip: 0
## nrows: 50000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant 21000
Processed variant 22000
Processed variant 23000
Processed variant 24000
Processed variant 25000
Processed variant 26000
Processed variant 27000
Processed variant 28000
Processed variant 29000
Processed variant 30000
Processed variant 31000
Processed variant 32000
Processed variant 33000
Processed variant 34000
Processed variant 35000
Processed variant 36000
Processed variant 37000
Processed variant 38000
Processed variant 39000
Processed variant 40000
Processed variant 41000
Processed variant 42000
Processed variant 43000
Processed variant 44000
Processed variant 45000
Processed variant 46000
Processed variant 47000
Processed variant 48000
Processed variant 49000
Processed variant 50000
Processed variant: 50000
## All variants processed## cutoff is specified, filtered vcfR object will be returned## 84.44% of SNPs fell below a completeness cutoff of 0.5 and were removed from the VCF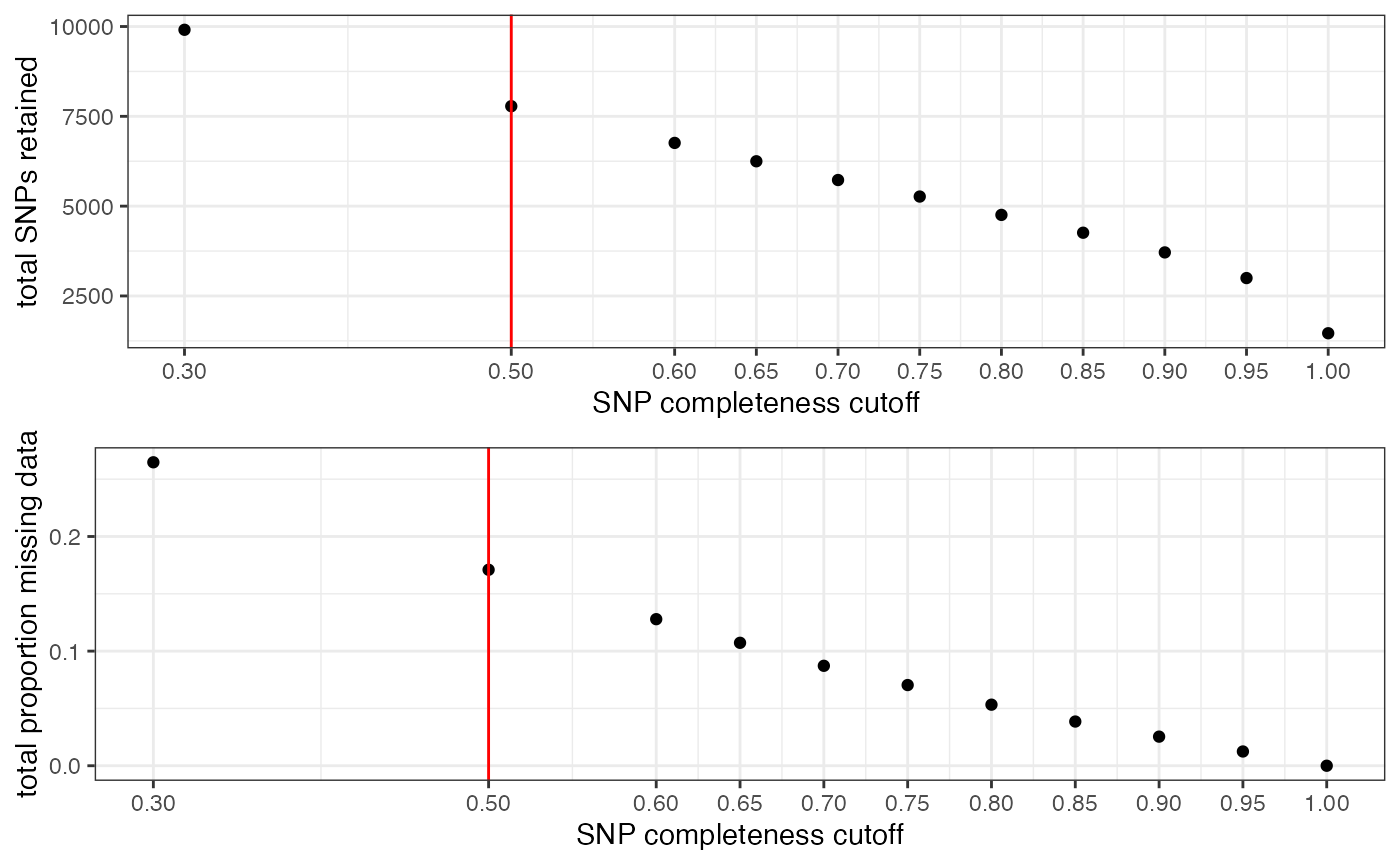
Filter each with VCFtools
cd /Users/devder/Desktop/benchmarking.vcfs
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.10K.vcf.gz --max-missing 0.5 --recode --recode-INFO-all --out 10K.filtered
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.20K.vcf.gz --max-missing 0.5 --recode --recode-INFO-all --out 20K.filtered
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.50K.vcf.gz --max-missing 0.5 --recode --recode-INFO-all --out 50K.filtered##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.10K.vcf.gz
## --recode-INFO-all
## --max-missing 0.5
## --out 10K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 1435 out of a possible 10000 Sites
## Run Time = 1.00 seconds
##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.20K.vcf.gz
## --recode-INFO-all
## --max-missing 0.5
## --out 20K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 3241 out of a possible 20000 Sites
## Run Time = 1.00 seconds
##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.50K.vcf.gz
## --recode-INFO-all
## --max-missing 0.5
## --out 50K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 7782 out of a possible 50000 Sites
## Run Time = 2.00 secondsRead in VCFtools files
#read in all the vcftools files
bash.10k<-read.vcfR("~/Desktop/benchmarking.vcfs/10k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 1435
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 1435
## Character matrix gt cols: 109
## skip: 0
## nrows: 1435
## row_num: 0
##
Processed variant 1000
Processed variant: 1435
## All variants processed
bash.20k<-read.vcfR("~/Desktop/benchmarking.vcfs/20k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 3241
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 3241
## Character matrix gt cols: 109
## skip: 0
## nrows: 3241
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant: 3241
## All variants processed
bash.50k<-read.vcfR("~/Desktop/benchmarking.vcfs/50k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 7782
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 7782
## Character matrix gt cols: 109
## skip: 0
## nrows: 7782
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant: 7782
## All variants processedValidate the min_mac() function
Filter each vcf with SNPfiltR
#perform filtering on each vcf file and save the result
r.10k<-min_mac(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.10K.vcf.gz"),min.mac = 2)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 10000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 10000
## Character matrix gt cols: 109
## skip: 0
## nrows: 10000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant: 10000
## All variants processed## 59.1% of SNPs fell below a minor allele count of 2 and were removed from the VCF
## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 20000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 20000
## Character matrix gt cols: 109
## skip: 0
## nrows: 20000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant: 20000
## All variants processed## 58.26% of SNPs fell below a minor allele count of 2 and were removed from the VCF
## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 50000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 50000
## Character matrix gt cols: 109
## skip: 0
## nrows: 50000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant 21000
Processed variant 22000
Processed variant 23000
Processed variant 24000
Processed variant 25000
Processed variant 26000
Processed variant 27000
Processed variant 28000
Processed variant 29000
Processed variant 30000
Processed variant 31000
Processed variant 32000
Processed variant 33000
Processed variant 34000
Processed variant 35000
Processed variant 36000
Processed variant 37000
Processed variant 38000
Processed variant 39000
Processed variant 40000
Processed variant 41000
Processed variant 42000
Processed variant 43000
Processed variant 44000
Processed variant 45000
Processed variant 46000
Processed variant 47000
Processed variant 48000
Processed variant 49000
Processed variant 50000
Processed variant: 50000
## All variants processed## 57.94% of SNPs fell below a minor allele count of 2 and were removed from the VCF
Filter each with VCFtools
cd /Users/devder/Desktop/benchmarking.vcfs
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.10K.vcf.gz --mac 2 --recode --recode-INFO-all --out 10K.filtered
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.20K.vcf.gz --mac 2 --recode --recode-INFO-all --out 20K.filtered
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.50K.vcf.gz --mac 2 --recode --recode-INFO-all --out 50K.filtered##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.10K.vcf.gz
## --recode-INFO-all
## --mac 2
## --out 10K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 4090 out of a possible 10000 Sites
## Run Time = 1.00 seconds
##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.20K.vcf.gz
## --recode-INFO-all
## --mac 2
## --out 20K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 8348 out of a possible 20000 Sites
## Run Time = 1.00 seconds
##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.50K.vcf.gz
## --recode-INFO-all
## --mac 2
## --out 50K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 21031 out of a possible 50000 Sites
## Run Time = 3.00 secondsRead in VCFtools files
#read in all the vcftools files
bash.10k<-read.vcfR("~/Desktop/benchmarking.vcfs/10k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 4090
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 4090
## Character matrix gt cols: 109
## skip: 0
## nrows: 4090
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant: 4090
## All variants processed
bash.20k<-read.vcfR("~/Desktop/benchmarking.vcfs/20k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 8348
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 8348
## Character matrix gt cols: 109
## skip: 0
## nrows: 8348
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant: 8348
## All variants processed
bash.50k<-read.vcfR("~/Desktop/benchmarking.vcfs/50k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 21031
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 21031
## Character matrix gt cols: 109
## skip: 0
## nrows: 21031
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant 21000
Processed variant: 21031
## All variants processedValidate the distance_thin() function
Filter each vcf with SNPfiltR
#perform filtering on each vcf file and save the result
r.10k<-distance_thin(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.10K.vcf.gz"),min.distance=100)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 10000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 10000
## Character matrix gt cols: 109
## skip: 0
## nrows: 10000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant: 10000
## All variants processed## 68 duplicated SNPs removed from input vcf##
|
| | 0%
|
|======================================================================| 100%## 2789 out of 9932 input SNPs were not located within 100 base-pairs of another SNP and were retained despite filtering
r.20k<-distance_thin(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.20K.vcf.gz"),min.distance=100)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 20000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 20000
## Character matrix gt cols: 109
## skip: 0
## nrows: 20000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant: 20000
## All variants processed## 143 duplicated SNPs removed from input vcf##
|
| | 0%
|
|======================= | 33%
|
|=============================================== | 67%
|
|======================================================================| 100%## 5608 out of 19857 input SNPs were not located within 100 base-pairs of another SNP and were retained despite filtering
r.50k<-distance_thin(vcfR = read.vcfR("~/Desktop/benchmarking.vcfs/benchmark.50K.vcf.gz"),min.distance=100)## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 50000
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 50000
## Character matrix gt cols: 109
## skip: 0
## nrows: 50000
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant 13000
Processed variant 14000
Processed variant 15000
Processed variant 16000
Processed variant 17000
Processed variant 18000
Processed variant 19000
Processed variant 20000
Processed variant 21000
Processed variant 22000
Processed variant 23000
Processed variant 24000
Processed variant 25000
Processed variant 26000
Processed variant 27000
Processed variant 28000
Processed variant 29000
Processed variant 30000
Processed variant 31000
Processed variant 32000
Processed variant 33000
Processed variant 34000
Processed variant 35000
Processed variant 36000
Processed variant 37000
Processed variant 38000
Processed variant 39000
Processed variant 40000
Processed variant 41000
Processed variant 42000
Processed variant 43000
Processed variant 44000
Processed variant 45000
Processed variant 46000
Processed variant 47000
Processed variant 48000
Processed variant 49000
Processed variant 50000
Processed variant: 50000
## All variants processed## 364 duplicated SNPs removed from input vcf##
|
| | 0%
|
|========== | 14%
|
|==================== | 29%
|
|============================== | 43%
|
|======================================== | 57%
|
|================================================== | 71%
|
|============================================================ | 86%
|
|======================================================================| 100%## 14439 out of 49636 input SNPs were not located within 100 base-pairs of another SNP and were retained despite filteringFilter each with VCFtools
cd /Users/devder/Desktop/benchmarking.vcfs
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.10K.vcf.gz --thin 100 --recode --recode-INFO-all --out 10K.filtered
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.20K.vcf.gz --thin 100 --recode --recode-INFO-all --out 20K.filtered
/Users/devder/Downloads/vcftools/src/cpp/vcftools --gzvcf benchmark.50K.vcf.gz --thin 100 --recode --recode-INFO-all --out 50K.filtered##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.10K.vcf.gz
## --recode-INFO-all
## --thin 100
## --out 10K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 2408 out of a possible 10000 Sites
## Run Time = 1.00 seconds
##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.20K.vcf.gz
## --recode-INFO-all
## --thin 100
## --out 20K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 4826 out of a possible 20000 Sites
## Run Time = 0.00 seconds
##
## VCFtools - 0.1.17
## (C) Adam Auton and Anthony Marcketta 2009
##
## Parameters as interpreted:
## --gzvcf benchmark.50K.vcf.gz
## --recode-INFO-all
## --thin 100
## --out 50K.filtered
## --recode
##
## Using zlib version: 1.2.11
## After filtering, kept 100 out of 100 Individuals
## Outputting VCF file...
## After filtering, kept 12541 out of a possible 50000 Sites
## Run Time = 1.00 secondsRead in VCFtools files
#read in all the vcftools files
bash.10k<-read.vcfR("~/Desktop/benchmarking.vcfs/10k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 2408
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 2408
## Character matrix gt cols: 109
## skip: 0
## nrows: 2408
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant: 2408
## All variants processed
bash.20k<-read.vcfR("~/Desktop/benchmarking.vcfs/20k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 4826
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 4826
## Character matrix gt cols: 109
## skip: 0
## nrows: 4826
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant: 4826
## All variants processed
bash.50k<-read.vcfR("~/Desktop/benchmarking.vcfs/50k.filtered.recode.vcf")## Scanning file to determine attributes.
## File attributes:
## meta lines: 14
## header_line: 15
## variant count: 12541
## column count: 109
##
Meta line 14 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 12541
## Character matrix gt cols: 109
## skip: 0
## nrows: 12541
## row_num: 0
##
Processed variant 1000
Processed variant 2000
Processed variant 3000
Processed variant 4000
Processed variant 5000
Processed variant 6000
Processed variant 7000
Processed variant 8000
Processed variant 9000
Processed variant 10000
Processed variant 11000
Processed variant 12000
Processed variant: 12541
## All variants processedcheck that every cell in the genotpye matrix matches for each file
#Does every cell in the genotype matrix for each camparable vcf match?
dim(r.10k@gt)## [1] 2789 101
dim(bash.10k@gt)## [1] 2408 101
dim(r.20k@gt)## [1] 5608 101
dim(bash.20k@gt)## [1] 4826 101
dim(r.50k@gt)## [1] 14439 101
dim(bash.50k@gt)## [1] 12541 101
#right away we can see that the dimensions of these vcf files don't match, indicating that the results differ.
#Let's calculate distance between SNPs in each vcf by hand to make sure both methods work as promised.
### Start with SNPfiltR thinned vcf:
##
#
#generate dataframe containing information for chromosome and bp locality of each SNP
df<-as.data.frame(r.10k@fix[,1:2])
#generate list of all unique chromosomes in alphabetical order
chroms<-levels(as.factor(df$CHROM))
#intialize empty df to hold filtering
keep.df<-data.frame()
#loop over each chromosome
#for t in vector containing the name of each chromosome
for (t in chroms){
#isolate the SNP positions on the given chromosome
fix.sub<-as.numeric(df$POS[df$CHROM == t])
#order the positions numerically
fix.sub<-fix.sub[order(fix.sub)]
#set the first position
prev<-fix.sub[1]
#initialize empty vector
k<-c()
k[1]<-NA
#loop to decide whether to keep each following SNP
if (length(fix.sub) < 2){
#if chrom only has 1 SNP, do nothing
} else{
#else, use a for loop to determine which SNPs to keep that satisfy our distance criteria
for (i in 2:length(fix.sub)){
#store distance in base pairs of the current SNP from the previous SNP
k[i]<- fix.sub[i] - prev
#update the current SNP to be the previous SNP for the next iteration of this loop
prev<-fix.sub[i]
#close for loop
}
#k[1]<- fix.sub[2] - fix.sub[1]
#close else statement
}
#make a dataframe with the precise info for each SNP for this chromosome
chrom.df<-data.frame(CHROM=rep(t, times=length(fix.sub)), POS=fix.sub, dist=k)
#now we rbind in the information for this chromosome to the overall df
keep.df<-rbind(keep.df,chrom.df)
#empty df for this chrom to prepare for the next one
chrom.df<-NULL
} #close for loop, start over on next chromosome
#order the dataframe to match the order of the input vcf file
order.df<-keep.df[match(paste(df$CHROM,df$POS), paste(keep.df$CHROM,keep.df$POS)),]
order.df<-order.df[order(order.df$dist),]
#show the lower distribution of SNP distances to see if any fall below the 100bp threshold
hist(order.df$dist, xlim =c(0,1000), breaks = 50000, main = "thinning using SNPfiltR",
xlab = "Distance from nearest SNP (base pairs)")
abline(v=100, col="red")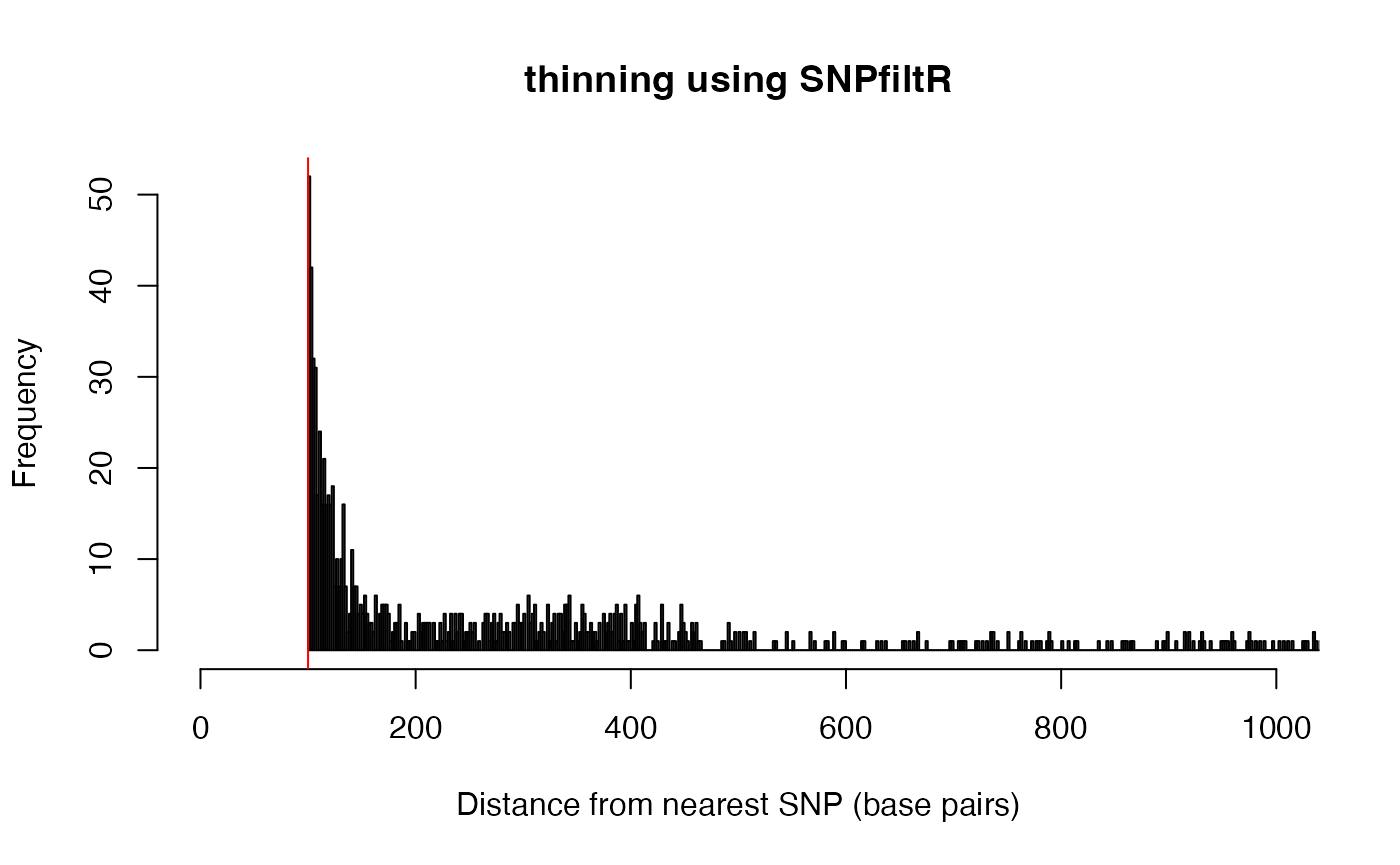
head(order.df) #show the tail of SNP distances## CHROM POS dist
## 220 Pseudo10_dna 2854287 101
## 331 Pseudo10_dna 3419169 101
## 333 Pseudo10_dna 3420934 101
## 467 Pseudo10_dna 4224139 101
## 469 Pseudo10_dna 4224491 101
## 532 Pseudo10_dna 4498157 101
#store df
snpfilt.df<-order.df
### repeat for the VCFtools filtered vcf
##
#
#generate dataframe containing information for chromosome and bp locality of each SNP
df<-as.data.frame(bash.10k@fix[,1:2])
#generate list of all unique chromosomes in alphabetical order
chroms<-levels(as.factor(df$CHROM))
#intialize empty df to hold filtering
keep.df<-data.frame()
#loop over each chromosome
#for t in vector containing the name of each chromosome
for (t in chroms){
#isolate the SNP positions on the given chromosome
fix.sub<-as.numeric(df$POS[df$CHROM == t])
#order the positions numerically
fix.sub<-fix.sub[order(fix.sub)]
#set the first position
prev<-fix.sub[1]
#initialize empty vector
k<-c()
k[1]<-NA
#loop to decide whether to keep each following SNP
if (length(fix.sub) < 2){
#if chrom only has 1 SNP, do nothing
} else{
#else, use a for loop to determine which SNPs to keep that satisfy our distance criteria
for (i in 2:length(fix.sub)){
#store distance in base pairs of the current SNP from the previous SNP
k[i]<- fix.sub[i] - prev
#update the current SNP to be the previous SNP for the next iteration of this loop
prev<-fix.sub[i]
#close for loop
}
#k[1]<- fix.sub[2] - fix.sub[1]
#close else statement
}
#make a dataframe with the precise info for each SNP for this chromosome
chrom.df<-data.frame(CHROM=rep(t, times=length(fix.sub)), POS=fix.sub, dist=k)
#now we rbind in the information for this chromosome to the overall df
keep.df<-rbind(keep.df,chrom.df)
#empty df for this chrom to prepare for the next one
chrom.df<-NULL
} #close for loop, start over on next chromosome
#order the dataframe to match the order of the input vcf file
order.df<-keep.df[match(paste(df$CHROM,df$POS), paste(keep.df$CHROM,keep.df$POS)),]
order.df<-order.df[order(order.df$dist),]
hist(order.df$dist, xlim =c(0,1000), breaks = 50000, main = "thinning using VCFtools",
xlab = "Distance from nearest SNP (base pairs)")
abline(v=100, col="red")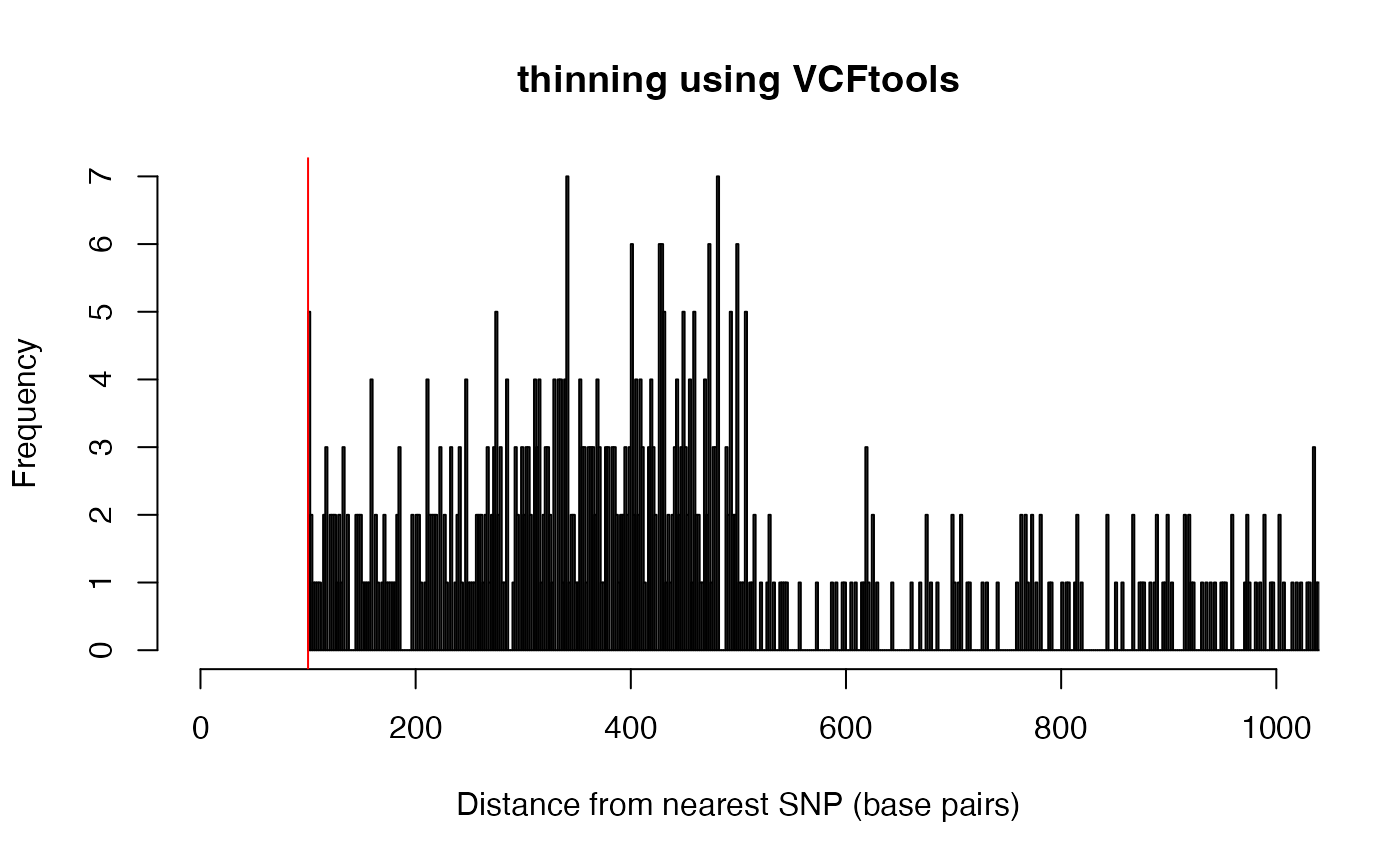
head(order.df) #show the tail of SNP distances## CHROM POS dist
## 2197 Pseudo10_dna 16289157 100
## 464 Pseudo10_dna 4498157 101
## 2257 Pseudo10_dna 16585988 101
## 163 Pseudo10_dna 2575297 102
## 1834 Pseudo10_dna 13584214 102
## 848 Pseudo10_dna 6642570 103
#putting the dataframe in chromosomal order, we can see right away that SNPs less than 100bp apart have been retained
order.df<-order.df[with(order.df, order(CHROM, POS)),]
order.df[1:10,]## CHROM POS dist
## 1 Pseudo10_dna 25891 NA
## 2 Pseudo10_dna 31885 5994
## 3 Pseudo10_dna 85182 53297
## 4 Pseudo10_dna 135731 50549
## 5 Pseudo10_dna 156488 20757
## 6 Pseudo10_dna 163096 6608
## 7 Pseudo10_dna 178396 15300
## 8 Pseudo10_dna 185542 7146
## 9 Pseudo10_dna 225731 40189
## 10 Pseudo10_dna 228849 3118
#plot the two together on the same histogram
order.df$approach<-rep("VCFtools", times=nrow(order.df))
snpfilt.df$approach<-rep("SNPfiltR", times=nrow(snpfilt.df))
sum.df<-rbind(order.df,snpfilt.df)
#plot both
ggplot(sum.df, aes(x=dist, color=approach)) +
geom_histogram(bins=100, fill="white")+
theme_classic()+
xlim(c(0, 1000))+
xlab(label = "Distance from nearest SNP (base pairs)")+
geom_vline(xintercept = 100, linetype="dashed")+
theme(legend.position = c(0.8, 0.8))## Warning: Removed 3756 rows containing non-finite values (`stat_bin()`).## Warning: Removed 4 rows containing missing values (`geom_bar()`).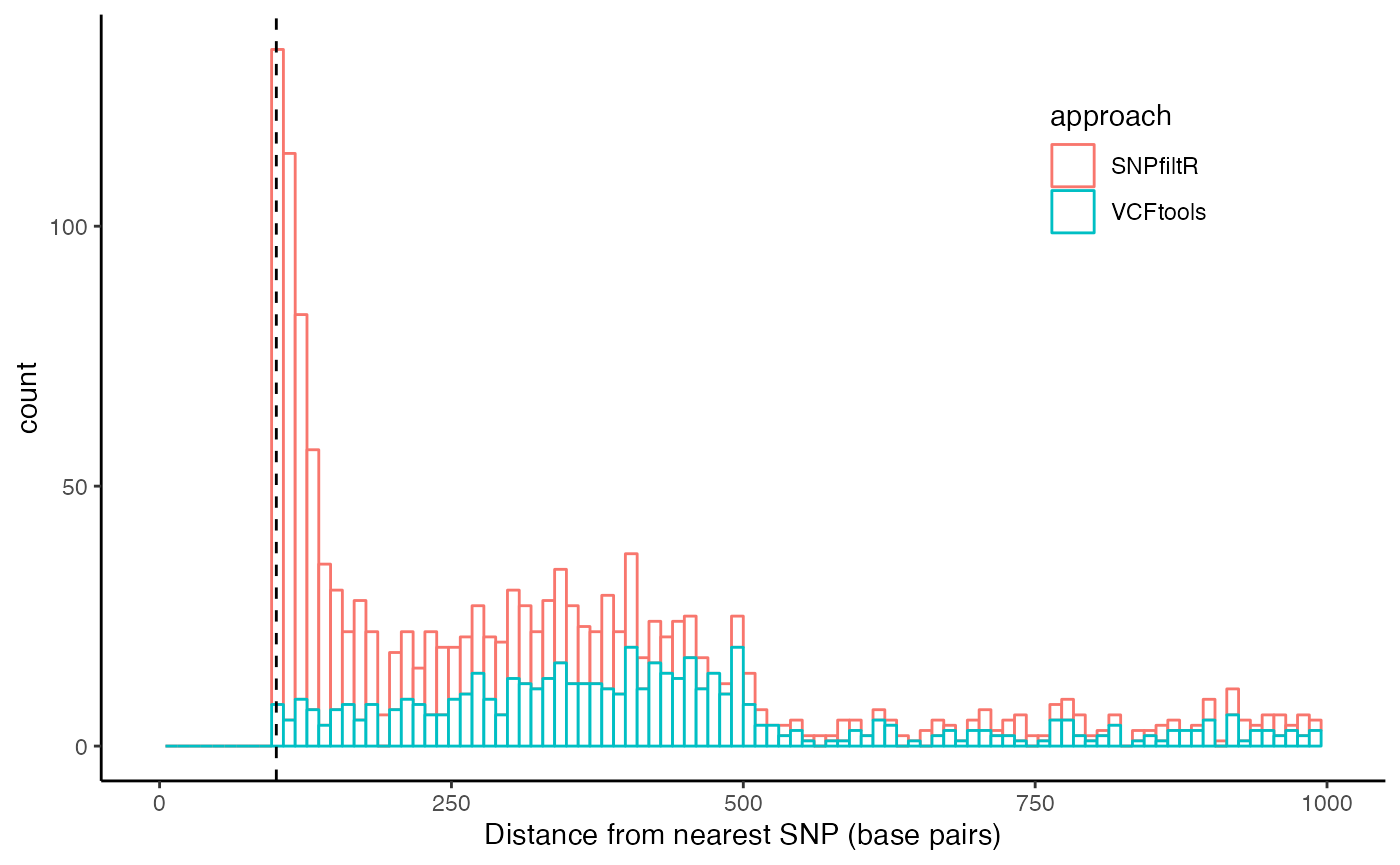
hists<-ggplot(sum.df, aes(x=dist, color=approach)) +
geom_histogram(bins=100, fill="white")+
theme_classic()+
xlim(c(0, 2500))+
xlab(label = "Distance from nearest SNP (base pairs)")+
geom_vline(xintercept = 100, linetype="dashed")+
theme(legend.position = c(0.8, 0.8))
#save plot
ggsave(hists,
filename = "~/Desktop/SNPfiltR.mol.ecol.resour.submission/resubmission/validate.thinning.pdf",
height = 2, width = 8, units = "in")## Warning: Removed 3061 rows containing non-finite values (`stat_bin()`).
## Removed 4 rows containing missing values (`geom_bar()`).
#see how many retained SNPs overlap between the two approaches for each file
table(with(as.data.frame(r.10k@fix), paste0(CHROM,POS)) %in% with(as.data.frame(bash.10k@fix), paste0(CHROM,POS)))##
## FALSE TRUE
## 1212 1577
table(with(as.data.frame(r.20k@fix), paste0(CHROM,POS)) %in% with(as.data.frame(bash.20k@fix), paste0(CHROM,POS)))##
## FALSE TRUE
## 2440 3168
table(with(as.data.frame(r.50k@fix), paste0(CHROM,POS)) %in% with(as.data.frame(bash.50k@fix), paste0(CHROM,POS)))##
## FALSE TRUE
## 6154 8285
#store number of SNPs for each approach as a vector
r.snps<-c()
r.snps[1]<-nrow(r.10k@gt)
r.snps[2]<-nrow(r.20k@gt)
r.snps[3]<-nrow(r.50k@gt)
#store number of bash SNPs
bash.snps<-c()
bash.snps[1]<-nrow(bash.10k@gt)
bash.snps[2]<-nrow(bash.20k@gt)
bash.snps[3]<-nrow(bash.50k@gt)
bar.frame<-data.frame(snps=c(r.snps,bash.snps),
approach=c("SNPfiltR::distance_thin()","SNPfiltR::distance_thin()","SNPfiltR::distance_thin()",
"VCFtools --thin","VCFtools --thin","VCFtools --thin"),
input.snps=c("10K","20K","50K",
"10K","20K","50K")
)
## Grouped bar plot
ggplot(bar.frame, aes(fill=approach, y=snps, x=input.snps)) +
geom_bar(position="dodge", stat="identity")+
xlab("SNPs in input vcf file")+
ylab("Number of thinned SNPs")+
theme_classic()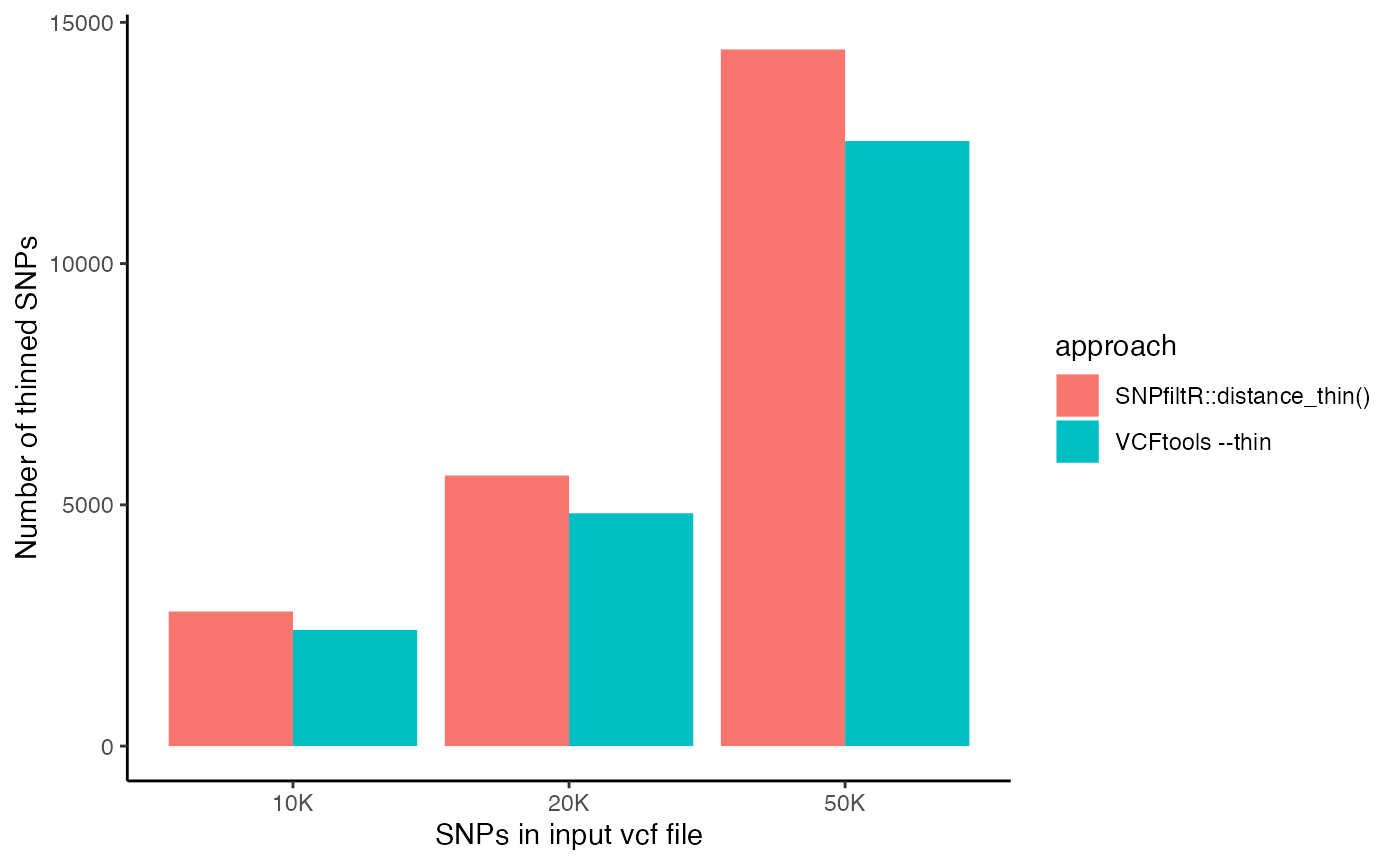
thin.bar<-ggplot(bar.frame, aes(fill=approach, y=snps, x=input.snps)) +
geom_bar(position="dodge", stat="identity")+
xlab("SNPs in input vcf file")+
ylab("Number of thinned SNPs")+
theme_classic()+
theme(legend.position = c(0.3, 0.8))
#save plot
ggsave(thin.bar,
filename = "~/Desktop/SNPfiltR.mol.ecol.resour.submission/resubmission/thinning.comp.pdf",
height = 3, width = 4.5, units = "in")Make final table showing genotpye concordance across comparisons
# print table showing concordance
concord<-data.frame(concordance=c(1,1,1,1,1,1,1,1,1,
sum(with(as.data.frame(r.10k@fix), paste0(CHROM,POS)) %in% with(as.data.frame(bash.10k@fix), paste0(CHROM,POS)))/nrow(as.data.frame(r.10k@fix)),
sum(with(as.data.frame(r.20k@fix), paste0(CHROM,POS)) %in% with(as.data.frame(bash.20k@fix), paste0(CHROM,POS)))/nrow(as.data.frame(r.20k@fix)),
sum(with(as.data.frame(r.50k@fix), paste0(CHROM,POS)) %in% with(as.data.frame(bash.50k@fix), paste0(CHROM,POS)))/nrow(as.data.frame(r.50k@fix))),
filtering=c("GQ","GQ","GQ","missing data","missing data","missing data",
"MAC","MAC","MAC","distance thin","distance thin","distance thin"),
input.snps=c("10K","20K","50K",
"10K","20K","50K",
"10K","20K","50K",
"10K","20K","50K")
)
#print table
heat <- reshape2::melt(concord)## Using filtering, input.snps as id variables
ggplot(data = heat, aes(x=input.snps, y=filtering, fill=value)) +
geom_tile(color = "white", size=1)+
geom_text(data=heat,aes(label=round(value, 2)))+
theme_minimal()+
scale_fill_gradient2(low = "white", high = "cornflowerblue", space = "Lab", name="genotype\nconcordance")+
ylab("type of filtering")+
xlab("Number of SNPs in input vcf file")## Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
## ℹ Please use `linewidth` instead.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.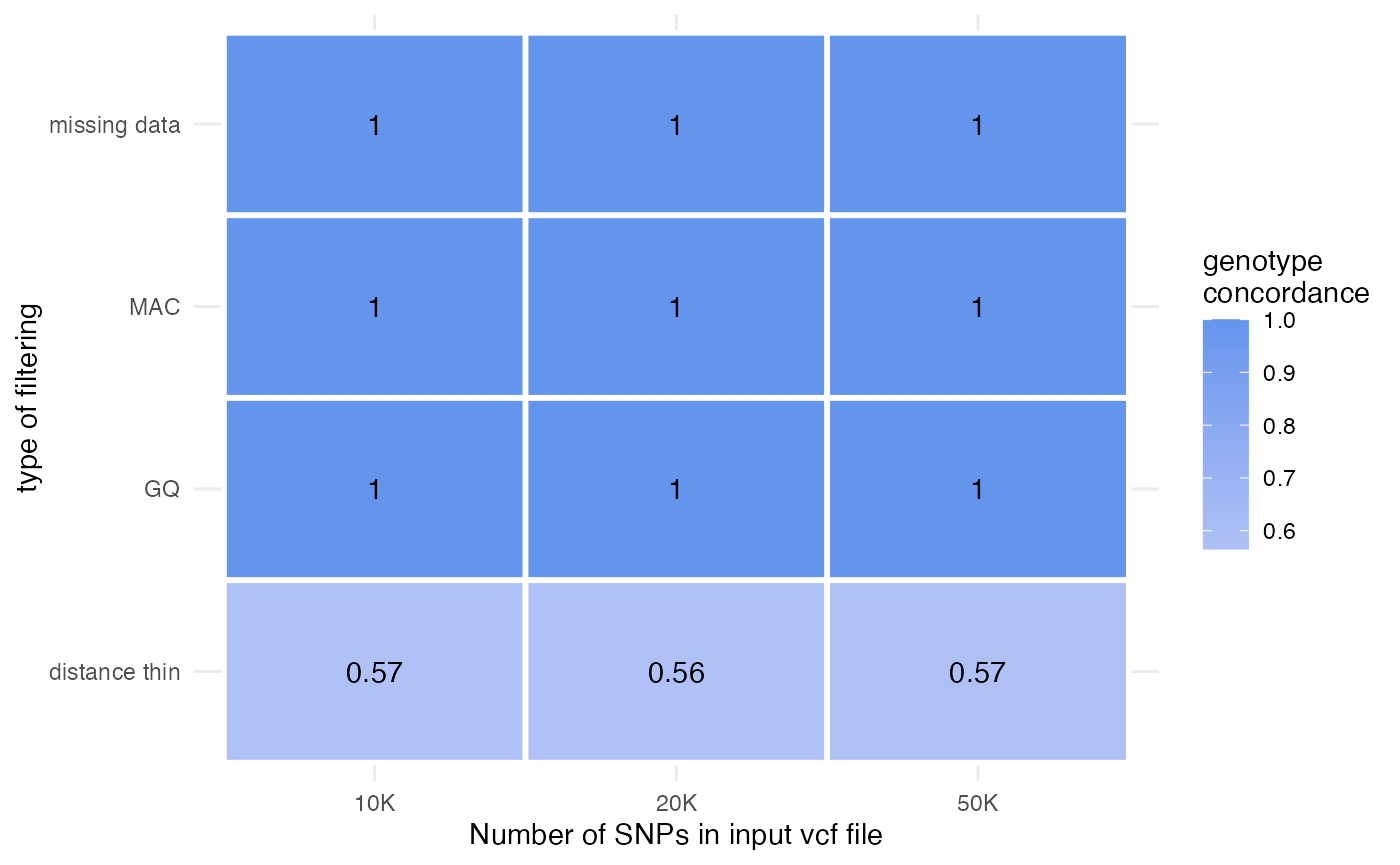
geno.concord.plot<-ggplot(data = heat, aes(x=input.snps, y=filtering, fill=value)) +
geom_tile(color = "white", size=1)+
geom_text(data=heat,aes(label=round(value, 2)))+
theme_minimal()+
scale_fill_gradient2(low = "white", high = "cornflowerblue", space = "Lab", name="genotype\nconcordance")+
ylab("type of filtering")+
xlab("Number of SNPs in input vcf file")
#save plot
#ggsave(geno.concord.plot,
# filename = "~/Desktop/SNPfiltR.mol.ecol.resour.submission/resubmission/geno.concord.pdf",
# height = 3, width = 5, units = "in")