Vizualise, filter based on Minor Allele Count (MAC)
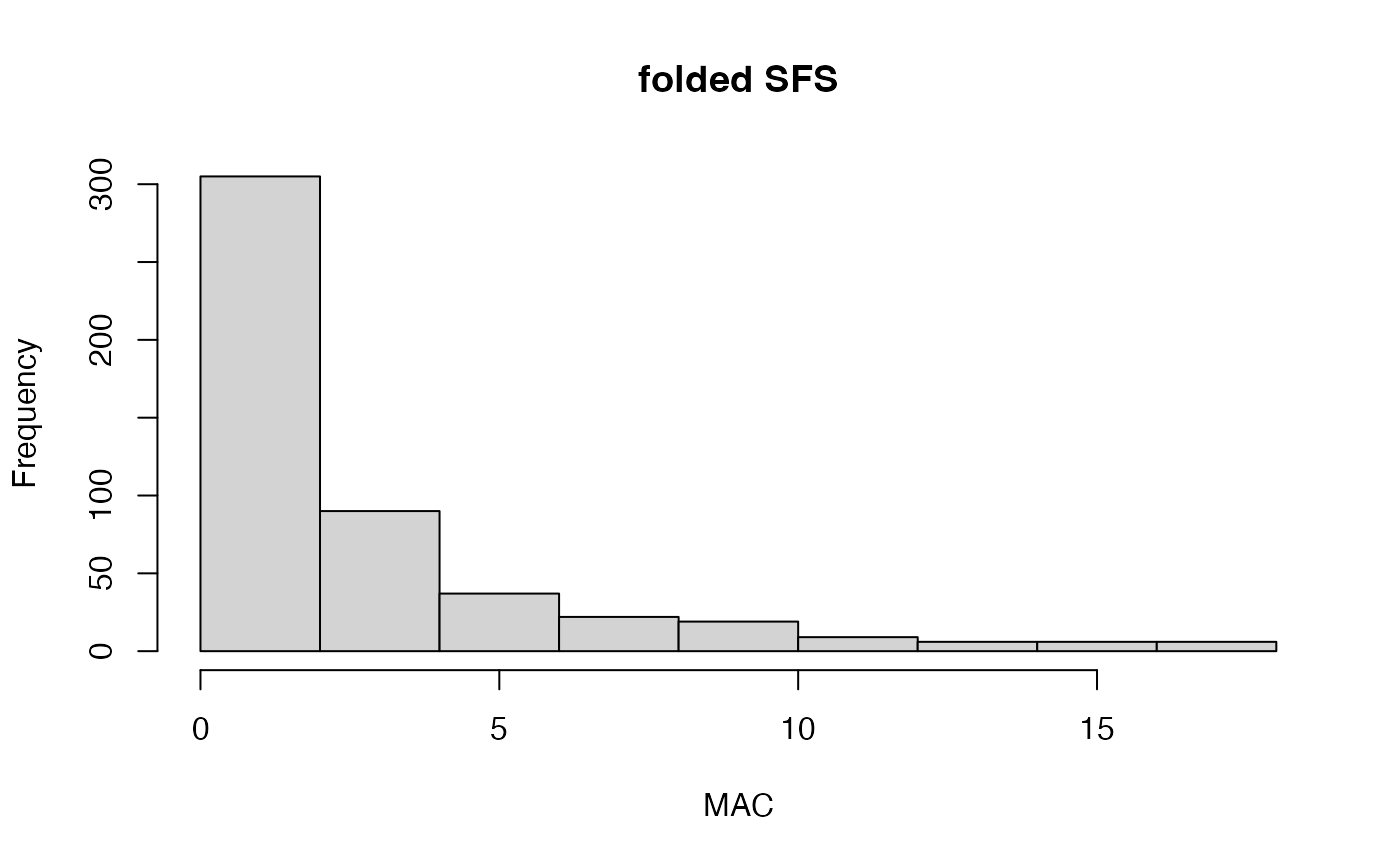
min_mac.RdThis function can be run in two ways: 1) Without 'min.mac' specified. This will return a folded site frequency spectrum (SFS), without performing any filtering on the vcf file. Or 2) With 'min.mac' specified. This will also print the folded SFS and show you where your specified min. MAC count falls. It will then return your vcfR object with SNPs falling below your min. MAC threshold removed. Note: previous filtering steps (especially removing samples) may have resulted in invariant SNPs (MAC =0). For this reason it's a good idea to run min_mac(vcfR, min.mac=1) before using a SNP dataset in downstream analyses.
min_mac(vcfR, min.mac = NULL)Arguments
- vcfR
a vcfR object
- min.mac
an integer specifying the minimum minor allele count for a SNP to be retained (e.g. 'min.mac=3' would remove all SNPs with a MAC of 2 or less)
Value
if 'min.mac' is not specified, the allele frequency spectrum is returned. If 'min.mac' is specified, SNPs falling below the MAC cutoff will be removed, and the filtered vcfR object will be returned.
Examples
min_mac(vcfR=SNPfiltR::vcfR.example)
#> no filtering cutoff provided, vcf will be returned unfiltered
 #> ***** Object of Class vcfR *****
#> 20 samples
#> 1 CHROMs
#> 500 variants
#> Object size: 0.7 Mb
#> 38.07 percent missing data
#> ***** ***** *****
#> ***** Object of Class vcfR *****
#> 20 samples
#> 1 CHROMs
#> 500 variants
#> Object size: 0.7 Mb
#> 38.07 percent missing data
#> ***** ***** *****