Remove SNPs with more than two alleles
filter_biallelic.RdThis function simply removes any SNPs from the vcf file which contains more than two alleles. Many downstream applications require SNPs to be biallelic, so this filter is generally a good idea during processing.
filter_biallelic(vcfR)Arguments
- vcfR
a vcfR object
Value
a vcfR object with SNPs containing more than two alleles removed
Examples
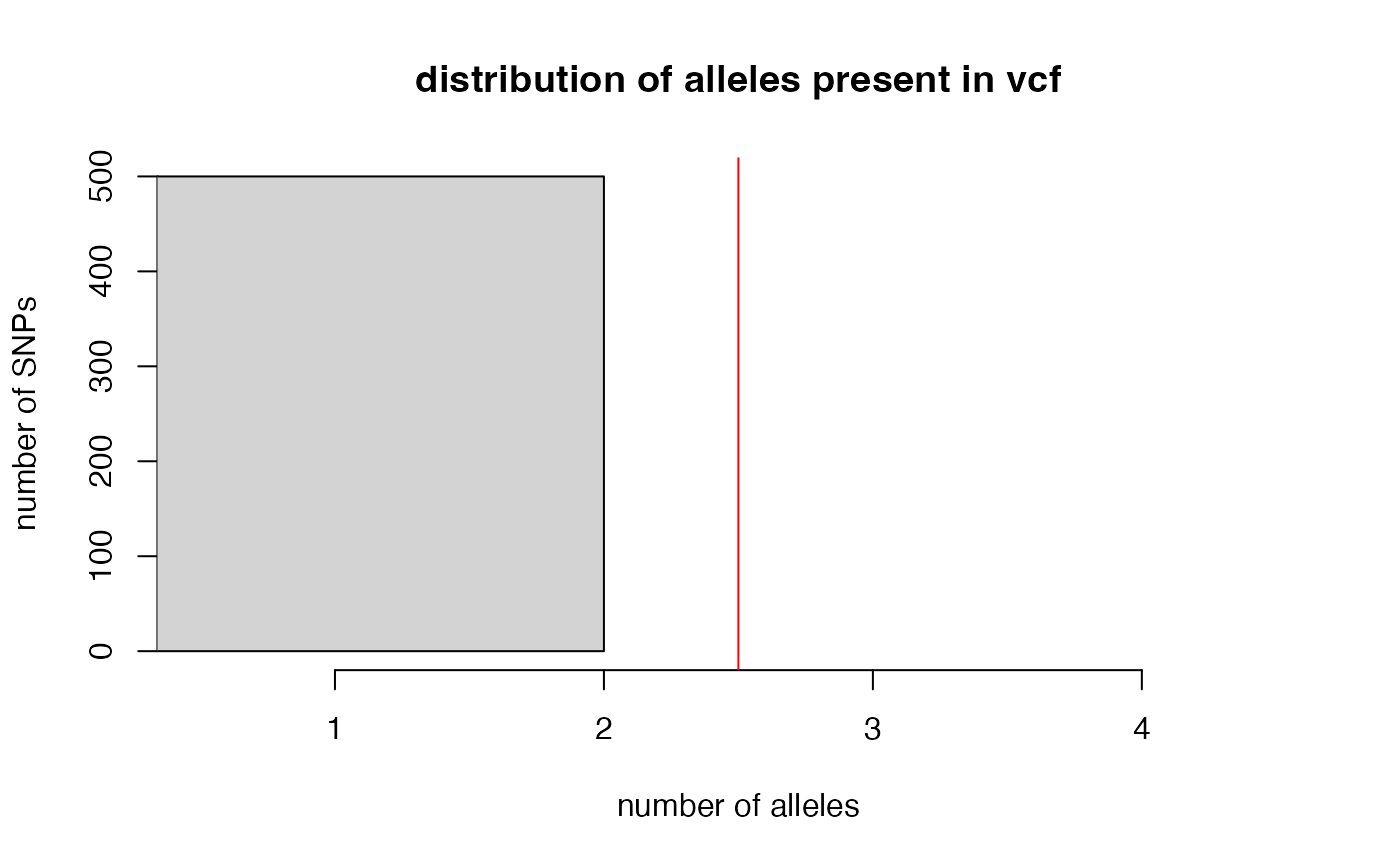
filter_biallelic(vcfR = SNPfiltR::vcfR.example)
#> 0 SNPs, 0% of all input SNPs, contained more than 2 alleles, and were removed from the VCF
 #> ***** Object of Class vcfR *****
#> 20 samples
#> 1 CHROMs
#> 500 variants
#> Object size: 0.7 Mb
#> 38.07 percent missing data
#> ***** ***** *****
#> ***** Object of Class vcfR *****
#> 20 samples
#> 1 CHROMs
#> 500 variants
#> Object size: 0.7 Mb
#> 38.07 percent missing data
#> ***** ***** *****