Filter out heterozygous genotypes failing an allele balance check
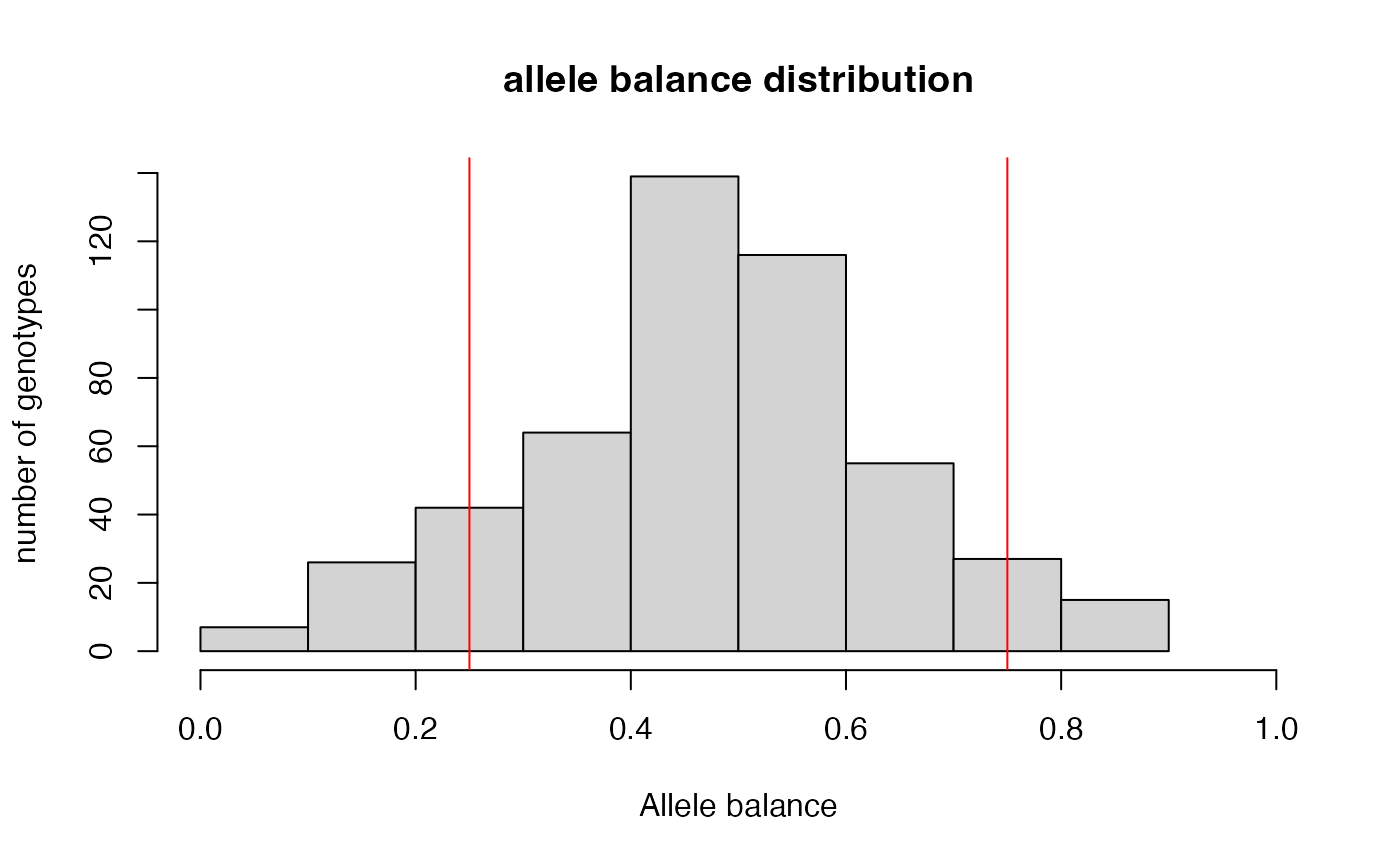
filter_allele_balance.RdThis function requires a vcfR object as input, and returns a vcfR object filtered to convert heterozygous sites with an allele balance falling outside of the specified ratio to 'NA'. If no ratio is specified, a default .25-.75 limit is imposed. From the dDocent filtering page "Allele balance: a number between 0 and 1 representing the ratio of reads showing the reference allele to all reads, considering only reads from individuals called as heterozygous, we expect that the allele balance in our data (for real loci) should be close to 0.5".
filter_allele_balance(vcfR, min.ratio = NULL, max.ratio = NULL)Arguments
- vcfR
a vcfR object
- min.ratio
minumum allele ratio for a called het
- max.ratio
maximum allele ratio for a called het
Value
An identical vcfR object, except that genotypes failing the allele balance filter have been converted to 'NA'.
Examples
filter_allele_balance(vcfR = SNPfiltR::vcfR.example)
#> 13.24% of het genotypes (1.05% of all genotypes) fall outside of 0.25 - 0.75 allele balance ratio and were converted to NA
 #> ***** Object of Class vcfR *****
#> 20 samples
#> 1 CHROMs
#> 500 variants
#> Object size: 0.7 Mb
#> 38.72 percent missing data
#> ***** ***** *****
#> ***** Object of Class vcfR *****
#> 20 samples
#> 1 CHROMs
#> 500 variants
#> Object size: 0.7 Mb
#> 38.72 percent missing data
#> ***** ***** *****