
My open science efforts
I believe that it is my responsibility as a scientist to make my work fully transparent and reproducible. I also believe that following the principles of transparency and reproducibility naturally lead to the creation of rigorous documentation, which can be viewed as a resource for training the next generation of scientists. Here I detail my special interest in creating resources that facilitate transparent and reproducible practices, AKA “open science”. Even this website is built from open-access resources found on GitHub and can be used as a template for others and my future self!
Publicly available DNA sequence data
To date, I have personally contributed 876 genomic samples from 39 different species to NCBI’s Sequence Read Archive (SRA; a publicly available repository for genomic sequence data) from my published manuscripts. Here is a detailed breakdown of those samples with a link to each BioProject:
| Project | Species | Samples | Sample Type |
|---|---|---|---|
| Scrub-jay species delimitation (PRJNA833486) | 4 | 115 | RADseq |
| Towhee Hybrid Zone (PRJNA915057) | 2 | 32 | Ultra-conserved Elements (UCEs) |
| Kingfisher mitonuclear discordance (PRJNA992577) | 8 | 83 | RADseq |
| Cyanolyca jay phylogeography (PRJNA996972) | 2 | 5 | Ultra-conserved Elements (UCEs) |
| Leaf-nosed bat phylogeography (PRJNA1084085) | 3 | 28 | RADseq |
| Identifying the source of introduced white-eyes (PRJNA1079333) | 6 | 124 | RADseq |
| Dwarf Kingfisher phylogeography (PRJNA1128088) | 10 | 135 | RADseq |
| Grosbeak Hybrid Zone (PRJNA1298969) | 2 | 156 | RADseq |
| Marsh Wren Hybrid Zone (PRJNA1330404) | 1 | 137 | RADseq |
| Flowerpecker phylogenomics (PRJNA1359933) | 1 | 60 | RADseq |
These publicly available sequence data can be freely used by anyone who wants to validate the results from my publications, or build on my work by incorporating any of these samples into their own future projects.
Building a codebase
By creating GitHub repositories containing detailed documentation for each of the first-author projects I’ve led, I have generated a large codebase containing helpful scripts for running a myriad of phylogenetic and population genetic analyses, which you can peruse here. In addition to making my individual projects transparent and reproducible, I take pride in maintaining this codebase as a public resource that can be used by the entire evolutionary genomics community. I am especially excited about the ability to use this codebase as a resource to help catalyze the growth of current and future mentees, helping to foster their rapid development into independent researchers.
Software
SNPfiltR
As part of my empirical work on the genomics of wild bird populations undertaken during my PhD, I identified a gap in available pipelines for analyzing next-generation sequence data (i.e., Illumina short-read sequences mapped to a reference genome). Specifically, there was no available software designed to filter single nucleotide polymorphism (SNP) datasets in an interactive way with built-in visualization tools. This typically resulted in users falling back on previously published sets of “standard filters”, rather than optimizing filtering parameters based on the idiosyncracies of their specific dataset. To address this gap I developed the R package SNPfiltR, which is designed to visualize key parameters such as genotype quality and missing data proportion, allowing users to quickly and interactively design an optimized set of filtering parameters for their SNP dataset, without the hassle and potential introduction of mistakes associated with designing unique, homebrewed visualization and filtering scripts for each SNP dataset. A manuscript announcing the package was published in Molecular Ecology Resources and the package is freely available for download via CRAN and GitHub. The package website https://devonderaad.github.io/SNPfiltR/ contains detailed tutorial-style vignette walkthroughs demonstrating the filtering process for real empirical datasets.
bgchm
I am involved as a co-author on the R package bgchm, which is focused on updating Bayesian approaches to genomic and geographic cline analysis using Hamiltonian Monte Carlo (HMC; as opposed to the traditional Markov Chain Monte Carlo, MCMC) for sampling posterior distributions. This update facilitates faster and more accurate sampling of posterior distributions, allowing the R package to handle genome-scale datasets. The manuscript announcing the details of the package update can be found here.
Workshops
RADseq analysis workshop
In Fall 2022 I led a group of graduate students through a semester-long weekly workshop detailing the process of analyzing RADseq data from start to finish. This workshop included hands-on training on: understanding file directory structure, navigating the command-line interface, using a high performance computing cluster (HPCC), using RStudio to interactively filter SNP datasets using SNPfiltR, and performing standard descriptive phylogenetic and population genetic analyses. The materials for this workshop are publicly available and documented in the following GitHub repository. Visualization of the workshop’s structure is here:
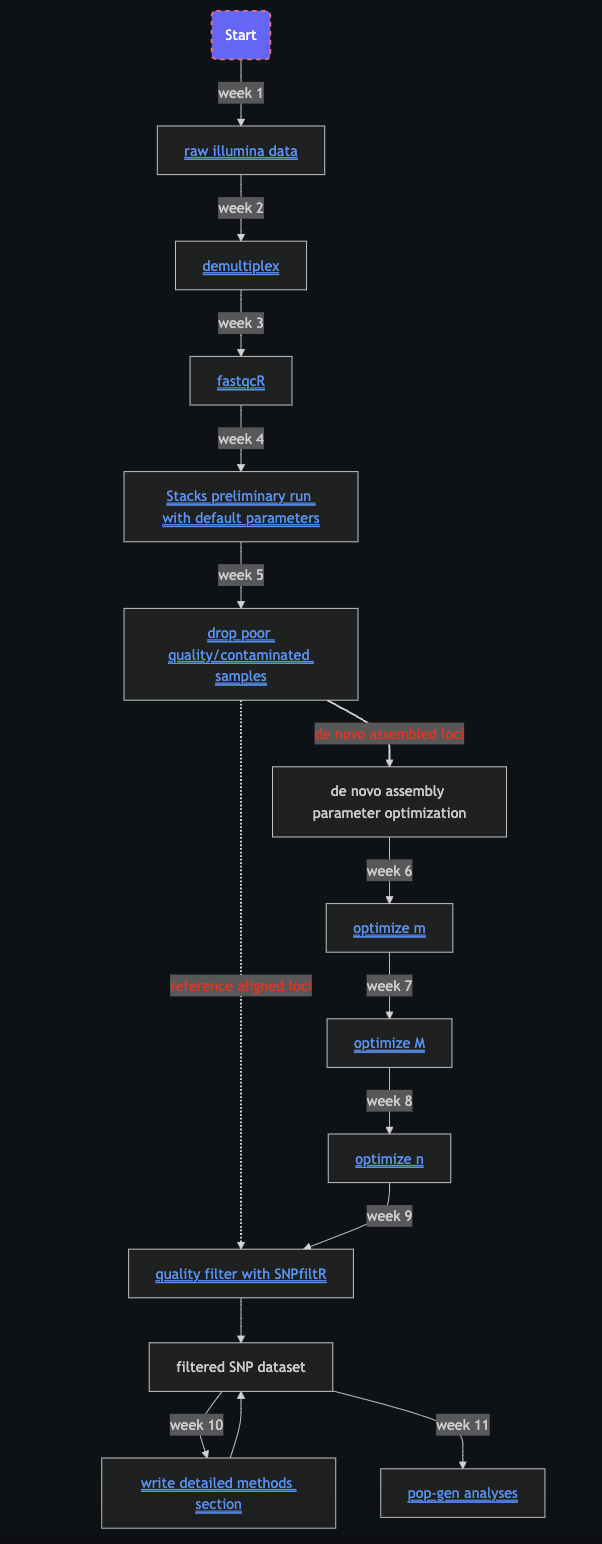
SNPfiltR workshop (AOS 2023)
At the American Ornithological Society meeting in Ontario, Canada (August 2023) I led a half-day workshop designed to take a group of investigators through the process of filtering their SNP datasets. The materials are based heavily on the SNPfiltR website and the exact details can be found in this publicly available GitHub repository.